痛风主要是由于嘌呤代谢紊乱和血液中尿酸慢性升高(高尿酸血症)引发的自发性炎症。当今社会中,人们对于高蛋白食品的摄入越来越多,这也导致高尿酸症的患病概率大幅升高,甚至发展成痛风。有证据显示痛风患者的肠道菌群与健康人群的肠道菌群结构存在差异,故本试验近一步探究痛风患者肠道菌群的具体结构。
试验设计
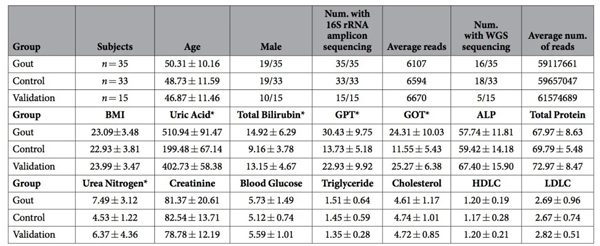
共选取83名志愿者参与试验,共分为三组。第一组为痛风组:由35名重度痛风患者组成;第二组为健康(对照)组:由33名健康人组成;第三组为验证组:由6名轻度痛风患者和9名健康人组成。对所有受试者的饮食以及基本情况等进行记录(表1),同时采集志愿者的血液样本和粪便样本,采用焦磷酸和Hiseq 2500相结合测序技术,对粪便样本的微生物多样性和功能基因组进行分析。
表1:合格受试者的基本情况详情
试验结果
1.痛风患者血液指标及肠道菌群变化
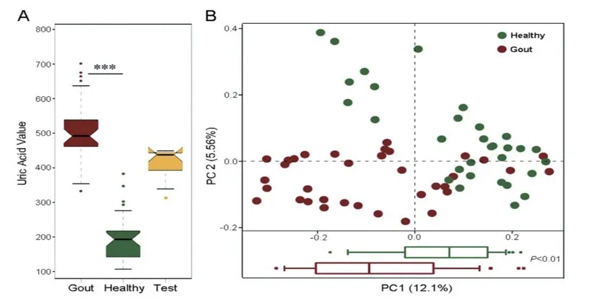
通过对16S rRNA基因进行测序分析,平均每个菌群共鉴定出202个OTU。在痛风组和健康组之间,没有发现年龄、性别或者BMI等方面的显著差异,但是在一系列血液指标中,发现两组之间的尿酸值、总胆红素谷氨酸-丙酮酸转氨酶(GPT)、谷氨酸草酰转氨酶(GOT)和尿素氮等水平存在显著差异(P<0.001;图1A)。
为了检测肠道菌群的微生物结构是否存在差异,基于属水平16S rRNA进行了加权UniFrac距离主坐标分析(PCoA),发现与健康组相比,痛风组的肠道菌群α多样性显著降低(P<0.01)。此外,痛风组和健康组肠道菌群的组织结构也存在高度不同,形成了两个簇,分别对应于两组(图1B),并且明显分离。
图1:三组志愿者尿酸水平以及主坐标分析详情
2.痛风的微生物指标
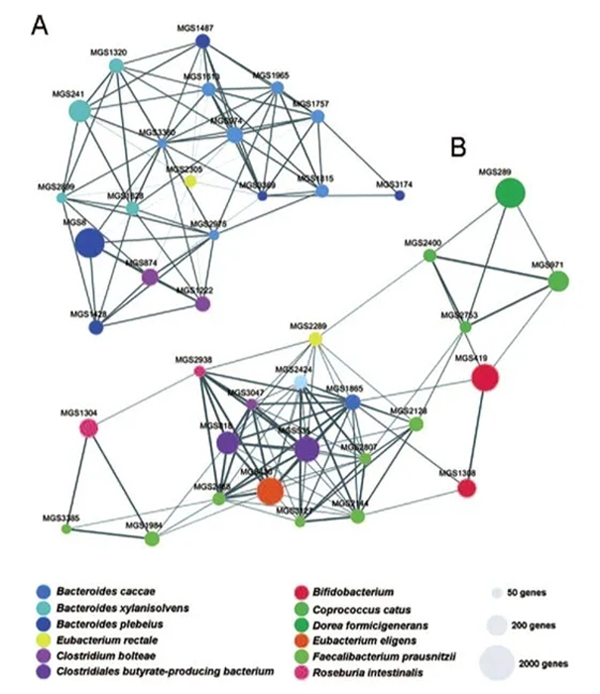
探索与痛风相关的微生物,根据基因组学数据从全基因组测序数据中预测微生物基因分为几类,每一类都被称为宏基因组种(MGS)。在样本中共鉴定出41种MGS,有22个富集于健康个体中,例如普氏杆菌、产丁酸梭菌和假双歧杆菌的MGS。另一方面,痛风患者中有19个MGS富集,包括Bacteroides caccae和Bacteroides xylanisolvens等(图2)。
同时基于16S rRNA焦磷酸测序,发现痛风组和健康组之间的细菌分布存在一定的相关性,共有17个属与痛风疾病相关,其中拟杆菌、Holdemania、Anaerotruncus等与痛风呈正相关,而Faecalibacterium、Coprococcus、Ruminococcus等与痛风呈负相关,与MGS分析确定的生物特征一致。
图2:肠道菌群分类学特征:A为痛风患者,B为健康个体
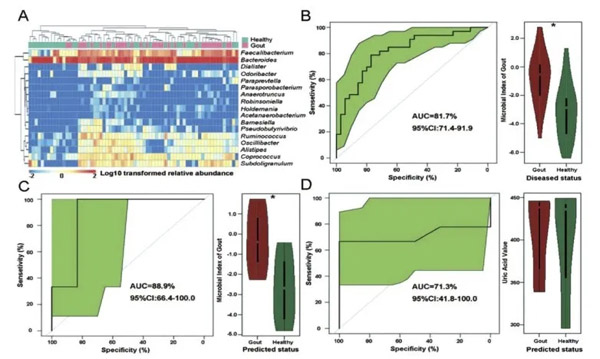
基于这17个差异菌属作为生物标记物评估痛风,并得出了“痛风微生物指数”(Mig),在标记物与受试者相关性热图显示,这些生物标记物可以区分健康个体和痛风患者(图3A)。同时痛风组和健康组之间的Mig明显不同(图3B),在ROC曲线中评估Mig模型,确定了Youden指数和Mig的阈值为-2.157,达到此阈值的Mig可能会增加患痛风的风险,这表明Mig可以用于高精度区分痛风个体。
为了验证该微生物模型的性能,对验证组的15名受试者作为试验对象,这15名受试者的血尿酸值高于对照组,但低于重度痛风患者的血尿酸值,但是血液指标并不是诊断痛风的唯一标准,经过6个月后的回诊,证实其中6人为痛风患者,9人为健康个体。对他们的肠道菌群分析显示,在验证组中基于Mig预测模型进行诊断的准确率达到88.9%(图3C),高于基于血尿酸为诊断标准的准确性(71.3%,图3D)。
图3:痛风相关微生物指标分析详情
3.痛风相关代谢途径分析
对痛风患者的相关功能代谢途径进行分析发现,有5245个KO和2286个COG与痛风呈相关性,将痛风相关的KOs映射到参考代谢途径上,痛风患者富含嘌呤、淀粉、蔗糖、鞘脂、丙氨酸、天冬氨酸、谷氨酸、视黄醇等相关代谢途径。在嘌呤代谢途径中,可将嘌呤降解为尿酸,同时生产尿素的尿囊素酶含量较健康人显著减少,无法进一步降解为尿素,从而导致痛风患者尿素水平的异常积累进而形成痛风。
试验结论
痛风患者在血液指标方面存在异常的同时,在肠道菌群方面也与健康个体之间存在显著差异,痛风患者的肠道微生物多样性显著低于健康个体。痛风患者体内缺少柔嫩梭菌和产丁酸梭菌,却富含Bacteroides caccae和Bacteroides xylanisolvens等。同时发现了17个属作为痛风生物标记物,可以高度区分痛风患者,痛风诊断准确率高。痛风患者肠道菌群合成嘌呤的能力减弱,而代谢嘌呤生成尿酸能力加强,同时生产尿素的尿囊素酶含量较健康人显著降低,最终造成尿酸堆积形成痛风。
 手机浏览
手机浏览