徐海燕 马慧敏 王彦杰 赵飞燕 刘亚华 海棠 张和平 孙志宏*[1]
(内蒙古农业大学乳品生物技术与工程教育部重点实验室,农业部奶制品加工重点实验室 呼和浩特 010018)
摘要 目的:研究长期高脂饮食为主的肥胖个体的肠道细菌多样性,比较不同健康状态的肥胖者肠道菌群的差异。方法:对14个肥胖个体根据健康状况分为3组(健康组,H组;胃肠道疾病组,D组;慢性病组,T组),提取3组样品粪便细菌宏基因组DNA,扩增其16S rRNA基因全长序列,利用三代测序技术测定样品宏基因组DNA的16S rRNA基因全长序列,采用相关生物信息学与统计学方法分析。结果:肥胖个体肠道细菌多样性降低,高丰度细菌含量增加,低丰度细菌含量减少,拟杆菌属(Bacteroides)是肥胖人群肠道细菌中含量最高的菌属。慢性病肥胖患者肠道菌群的B/F值(拟杆菌门/厚壁菌门,Bacteroidetes/Firmicutes)降低,同时产气柯林斯菌和Lachnospira peotinoschiza的相对含量显著高于健康组的肥胖个体(LDA>2)。此外,患有腹泻等胃肠道疾病的肥胖个体(D组)变形菌门下的一些致病菌相对含量增加。结论:肥胖个体的健康状态影响其肠道菌群结构和组成,肥胖引发的不同并发症可能与肠道菌群相关。
关键字 肥胖;肠道细菌;高脂饮食;三代测序
The Study on Intestinal Bacterial Diversity in Obese Individuals
Xu Haiyan Ma Huimin Wang Yanjie Zhao Feiyan Liu Yahua Hai Tang Zhang Heping Sun Zhihong*
(Key Laboratory of Dairy Biotechnology and Engineering Ministry of Education, Inner Mongolia Agricultural University, Key Laboratory of Dairy Products Processing,Ministry of Agriculture, Hohhot 010018)
Abstract Objective: The intestinal bacterial diversity of 14 high-fat diet-induced obese individuals under different health was investigated and the difference of intestinal bacteria from obese individuals of different health condition was compared. Method: 14 obese people were divided into three group according to their health condition (Health group, H group; gut disease, D group and chronic disease, T group). The DNA was extracted from the stool samples in the 3 groups and the full length 16S rRNA gene sequences were amplified, then the products of PCR were sequencing using Pacific Biosciences single molecule real-time sequencing technology (PacBio SMRT sequencing). Results: The intestinal bacterial diversity of obese people was decreased, Bacteroidetes and Firmicutes were more abundant, but the low abundance bacteria were more less, and Bacteroides had the highest proportion in core bacteria. The ratio of the relative abundance of Bacteroidetes to that of Firmicutes (B/F ratio) was lower in chronic disease obese group than health group, and the relative abundance of Collinsella aerofaciens and Lachnospira peotinoschiza was significantly higher than obese individuals in health group. In addition, The Parabacteroides was higher in obese individuals of gut disease group. Conclusion: The health condition of obese individuals had an effect on the structure and composition of intestinal bacteria, the complications of obesity may be associated with the changes of intestinal microbial population.
Keywords obesity;intestinal bacteria;high-fat diet;PacBio SMRT sequencing
人体肠道内栖息着一个独特的微生物群体,包括细菌、真菌、古菌和病毒。所有这些微生物的基因组数量超过人类基因组100倍以上,其中单细菌的种类就在千种以上,数量达到1014个,数量庞大,群类结构复杂[1]。过去的几年里,肠道菌群已被当作人体的一个“新器官”,其重要性为越来越多的科学家所重视。已经证实肠道微生物与机体相互作用,影响着宿主整体的健康,在食物消化和能量转运,免疫系统调控,基因表达,神经系统作用等方面都扮演着重要的角色[2]。而肠道微生物的种类、数量、分布及其功能也受多种因素的影响,包括生活方式、饮食习惯、卫生条件和药物等[3]。鉴于肠道菌群与宿主健康息息相关,如何维持肠道菌群健康和稳定已成为科学家研究的重点。
近年来,不依赖于纯培养的高通量测序技术的兴起,使人们对肠道微生物及其基因组有了更深入的了解,借助新的测序技术,巨大而复杂的微生物群落以及它们和宿主间的互作机制逐渐被发现。覃俊杰等[4]利用高通量测序技术研究了中国人II型糖尿病患者的肠道菌群,发现患者肠道菌群严重失调,特别是丁酸盐产生细菌肠道罗伊氏菌(Roseburia intestinals)和柔嫩梭菌(Faecalibacterium prausnitzii)显著减少。华盛顿大学Jeffrey Gordon团队早期的研究已经证明,肥胖和肠道微生物的改变有关,且肥胖会导致菌群多样性降低[5]。随后他们用无菌小鼠模型证实了这一点,并发现了菌群在肥胖发生过程中的作用[6]。
由于肠道菌群最重要,也是最基本的作用是获取能量,因此饮食习惯很大程度上影响肠道菌群的结构和组成。对两例健康个体肠道菌群元基因组的测序结果发现:肠道细菌富含可以代谢淀粉、蔗糖、甘露糖、葡萄糖、阿拉伯糖和木糖等至少81种糖基水解酶的基因家族,这些基因大部分是人类自身所不具备的。肠道菌群代谢食物中的植物纤维是多种微生物共同协作的结果[7]。2014年的一项研究表明,高脂的动物性食品(如:肉、蛋、奶等)比植物性食品(如:水果和蔬菜等)对肠道菌群的影响更大,在食用动物性食品人群的肠道中,耐胆盐的细菌如Alistipes、Bilophila(嗜胆菌属)和Bacteroides(拟杆菌属)显著增加,而代谢植物多糖的Firmicutes(厚壁菌门)显著减少[8]。虽已有大量研究提示高脂饮食引起肥胖的发生是由于肠道菌群的改变,但绝大多数研究还局限在动物模型上,对于人群的研究结果报道不一。例如对12例肥胖个体的研究[9]发现,肥胖者肠道菌群在门的水平上发生改变,而另一个23例肥胖者的研究[10],却没有发现相同的结果。由于肥胖是II型糖尿病、高血压、酒精性脂肪肝等多种疾病的主要因素[11],且高脂饮食引发的肥胖患者的肠道菌群结构和组成是怎样的,哪些肠道细菌参与了疾病的发生尚未有统一的定论,因此研究高脂饮食引发的肥胖人群肠道菌群仍具有现实意义。
本研究募集14个肥胖志愿者,日常饮食均以高脂的动物食品为主,利用SMRT RS II 三代测序技术以及相关生物信息学与统计学分析方法分析其肠道菌群的多样性,同时比较不同健康状态下肥胖个体的肠道菌群的结构和组成差异,旨在为高脂饮食引发的肥胖人群肠道菌群多样性数据库添加新的数据。
1. 材料与方法
1.1材料与试剂
1.1.1试验对象 本研究对象来自中国内蒙古呼和浩特市,14位志愿者年龄在40~60岁之间,长期以肉类等高脂饮食为主,体质指数(BMI)为25.03~35.83 kg/m2(世界卫生组织西太平洋地区官员、国际肥胖研究协会和国际肥胖工作组2000年2月联合发布的《亚太区肥胖的重新定义和处理》把BMI≥25 kg/m2定义为肥胖。)其中按照身体健康状况分为3组,D组包括5位近期有如腹泻等胃肠道疾病的5位志愿者,H组是尚未有疾病表现的5位志愿者,T组的4位志愿者有高血糖或者高血压、脂肪肝等慢性病史。所有参与者在采样前3个月内未服用过任何抗生素或益生菌。人体样本的使用经过内蒙古农业大学认可,所有受试者均表示同意参加本试验,具体信息见表1。
表1 实验对象的信息
Table 1 The information of experimental subjects
|
编号 |
分组 |
性别 |
年龄 |
身高/m |
体重/kg |
BMI |
疾病史 |
|
1 |
D |
女 |
43 |
1.58 |
71 |
28.44 |
腹泻 |
|
2 |
D |
男 |
56 |
1.71 |
83 |
28.38 |
肠胃炎 |
|
3 |
D |
男 |
48 |
1.76 |
85 |
27.44 |
肠胃炎 |
|
4 |
D |
男 |
52 |
1.72 |
87.5 |
29.58 |
结肠炎 |
|
5 |
D |
男 |
43 |
1.65 |
68.5 |
25.16 |
腹泻 |
|
6 |
H |
男 |
53 |
1.68 |
79 |
27.99 |
无 |
|
7 |
H |
男 |
46 |
1.67 |
78 |
27.97 |
无 |
|
8 |
H |
男 |
52 |
1.68 |
75 |
26.57 |
无 |
|
9 |
H |
女 |
41 |
1.62 |
67 |
25.53 |
无 |
|
10 |
H |
男 |
57 |
1.68 |
76 |
26.93 |
无 |
|
11 |
T |
女 |
45 |
1.66 |
69 |
25.04 |
高血压 |
|
12 |
T |
男 |
51 |
1.72 |
106 |
35.83 |
高血压 |
|
13 |
T |
男 |
60 |
1.69 |
71.5 |
25.03 |
糖尿病 |
|
14 |
T |
男 |
54 |
1.73 |
82 |
27.40 |
高血压、脂肪肝 |
1.1.2试剂 MO BIO Power Fecal® DNA Isolation Kit试剂盒,美国Mo Bio公司;Taq DNA聚合酶、dNTP mix、10x Easy Taq Buffer等PCR体系试剂,大连宝生物工程有限公司;5×TBE电泳缓冲液(Tris碱54 g、Na2EDTA·2H2O 3.72 g、硼酸 27.5 g,定容至1000 mL,pH 8.0),1.0%的琼脂糖胶(1.0 g琼脂糖溶于100 mL 0.5×TBE缓冲液),天津基准化学试剂公司;SMRT 建库试剂盒(DNA Template Prep Kit 1.0)、测序试剂(DNA/Polymerase Bingding Kit P6 v2),美国太平洋生物科学公司。
1.2仪器与设备
SMRT RS II 测序平台,美国Pacific Biosciences公司;ML-30L型全自动高压蒸汽灭菌器,日本三洋公司;Applied Biosystems PCR仪,美国应用生物系统公司;ND-1000 型微量紫外分光光度计,美国赛默飞科技公司;HWS24 型恒温水浴锅,上海一恒科学仪器有限公司;CDS8000型UPV凝胶成像分析系统,上海赛智创业科学公司; Eppendorf 5810R高速冷冻离心机,德国艾本德公司;Qubit 2.0,美国生命技术有限公司。
1.3方法
1.3.1样品采集 所有志愿者采集1次新鲜粪便样品。每个样品采集均由志愿者个人完成,用试验组提供的无酶无菌管采集约15 g的新鲜粪便样品,样品采集后立即加入15 mL的保护剂,迅速冷冻,干冰冻藏寄回内蒙古农业大学乳品生物技术与工程教育部重点实验室,立即进行预处理并提取DNA。
1.3.2粪便细菌宏基因组DNA提取 取0.3g充分混匀的粪便样品,采用Power Fecal® DNA Isolation Kit试剂盒,依照说明书的提取步骤抽提样品中细菌宏基因组DNA,用1.0%琼脂糖凝胶电泳和微量紫外分光光度计检验DNA样品的完整度、纯度以及浓度,将符合后续试验要求的DNA置于-80 ℃冰箱,备用。
1.3.3细菌16S rRNA基因全长扩增14个样品的细菌宏基因组DNA16S rRNA基因全长序列被扩增,扩增引物为27F(5’-AGAGTTTGATCMTGGCTCAG-3’)和1492R(5’-ACCTTGTTACGACTT-3’)[12]。因为测序时14个样品需要按照等质量混样后构建在同一个文库里,所以为了区分同一个文库中的不同样品,用带有识别标签(Barcode)的引物对每个样品进行PCR扩增,以区分每个样品。将1.3.2节提取的DNA作为模板,用上述引物进行PCR扩增,每个样品保证是独特的标签引物,PCR程序设置为:95℃预变性2min;95℃变性30s,55℃退火45s,72℃延伸1min,共循环28次;72℃终端延伸7min,4℃终止。具体扩增体系如表2所示。
表2聚合酶链式反应扩增条件
Table 2 The condition of polymerase chain reaction amplification
|
体系 |
剂量/μL |
|
10×PCR buffer |
5 |
|
dNTP MIX(2.5mmol/L each) |
4 |
|
正向引物(10pmol/μL) |
1.0 |
|
反向引物(10pmol/μL) |
1.0 |
|
基因组DNA模板(100 ng/μL) |
1.5 |
|
TaKaRa Taq酶(5U/μL) |
0.5 |
|
ddH2O |
补充至50μL |
1.3.4文库构建和PacBio SMRT三代测序 每个样品的PCR产物经纯化后等质量混合,用试剂盒构建文库,根据厂商提供的说明书严格执行。将构建好的文库,加测序引物和聚合酶后,使用PacBio RS II仪器上机测序。
1.3.5高质量序列的提取 使用RS_ReadsOfinsert.1对原始序列进行质控,只保留符合以下标准的序列:①插入片段重复测序的次数≥5;②最小预测精确度为90%;③最短插入序列长度为1400 nt;④最长序列长度为1800 nt。符合以上标准的序列定义为高质量的序列,并纳入下一步的生物信息学分析。
1.3.6生物信息学分析 根据标记的Barcode将原始序列划分到每个样品中,然后去掉Barcode和引物序列,应用QIIME平台(v1.70)对高质量的序列进行生物信息学分析。主要步骤如下:
1)PyNAST校准并把序列排齐,在100%相似性下进行UCLUST归并,建立无重复的16S rRNA基因全长序列集。
2)将所有序列进一步在98.65%相似性下聚类获得分类操作单元 (Operational taxonomic units,OTU)[13]。
3)应用ChimeraSlayer去除含有嵌合体序列的OTU序列;
4)使用RDP(Ribosomal Database Project)和Greengenes(Version _3.8)数据库进行比对,对去除嵌合体后的OTU进行序列同源性比对和分类学的门、纲、目、科、属和种水平上进行鉴定。
5)使用FastTree软件,绘制基于OTU代表序列的系统发育进化树。随后,计算α和β多样性。采用香农指数(Shannon-Wiener index)、获得菌种数量值(Observed species)、超1指数(Chao1 index)和辛普森指数(Simpson index)共4个α多样性指标,分别对样品菌群构成的丰度和多样性进行评价。同时,采用稀疏曲线(Rarefaction curve)和香农多样性指数(Shannon’s diversity index)评估每个样本测序的多样性,以及评估测序量和各个文库的物种多样性和丰富。
6)基于加权(Weighted)和非加权(Unweighted)主坐标分析(Principal coordinate analysis,PCoA)的UniFrac距离的非加权组平均法(Unweighted pair-group method with arithmetic means,UPGMA)聚类分析对不同样品间微生物群落结构差异进行计算。
1.3.7数据处理 首先,通过维恩(Venn)图展现两组样品中所共有的OTU 数目,采用LEfSe基于线性判别分析(Linear discriminant analysis, LDA)效应量(Effect size)的分析方法,将非参数统计检验(Kruskal-Wallis 多组检验与 Wilcoxon 秩和检验)与线性判别分析相结合,检测不同组样品中微生物的门、属和种水平上的差异,采用Origin 8.6 软件、Graphpad 6.0和R语言软件(v3.3.2)作图。
1.4核酸序列登录号
本试验所有测序数据上传到MG-RAST数据库,项目检索号为:mgp83562。
2结果与分析
2.1 测序丰度评估和α多样性分析
测序后经过序列过滤,14个样品共获得了134453条高质量的16S rRNA基因序列,平均每个样品序列的数为9604条(范围=2257-26359,SD=6521)。共划分得到28833个OTUs进行下一步分析,平均每个样品OTU的数量为2438个(范围=393-7354,SD=1861)。为评估测量是否满足后续分析,绘制稀疏曲线和香浓曲线,其中,稀疏曲线未进入平台期,说明随着测序量的增加,有可能会有新的种系被发现;香浓曲线已接近平台期,表明虽然随着测序深度的增加,有可能发现新的物种,但当前测序量已可覆盖样品中大部分的微生物组成,满足后续分析要求(图1 a和1b)。
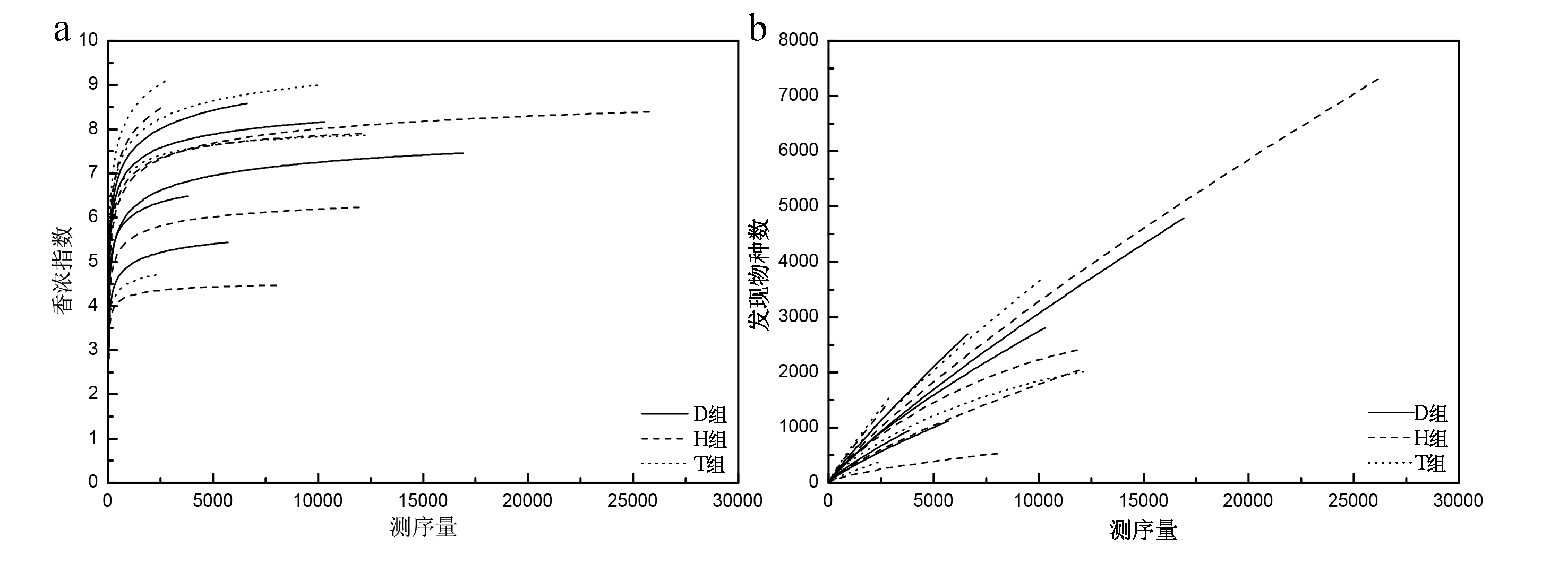
图1样品中细菌群体的香浓曲线(a)和稀疏曲线(b)
Fig.1 Shannon diversity (a) and Rarefaction (b) curves of bacterial populations in samples
为评估肥胖人群肠道微生物的物种多样性和丰度,计算物种多样性的香浓指数和辛普森指数以及评价物种丰度的超1指数和发现物种数(表3),其中香浓指数均值6.89(测序深度为2557条序列数),然而,基于三代测序技术测定人体肠道微生物的α多样性值,测序深度在2012条序列数时,健康人体肠道菌群的香浓指数均值大于8[14],由此推测长期以肉类等高脂饮食为主的肥胖个体肠道菌群物种多样性低于正常体重的个体,这和2009年Turnbaugh等[5]的研究结果一致。此外,按照不同健康状况将14个人的粪便样品分为3组,即健康组(H组)、胃肠病组(D组)和慢性病组(T组),3组样品的α多样性指数没有统计学上的显著性差异(P>0.05,Mann-Whitney U检验),说明不同健康状态的肥胖患者的菌群多样性和丰度并没有因健康状态不同而存在差别,这或许是因为14个研究对象饮食都是高脂饮食的缘故。研究已证实,饮食是塑造肠道菌群的一个重要因素[8]。
表3测序深度和α多样性指数的信息
Table 3 The information of sequencing depth and α diversity index
|
样品编号 |
序列数 |
OTU数目 |
超1指数 |
发现物种数 |
香浓指数 |
辛普森指数 |
|
1 |
10396 |
2828 |
4313.83 |
912.13 |
7.58 |
0.97 |
|
2 |
5762 |
1126 |
3243.75 |
566.15 |
5.21 |
0.82 |
|
3 |
6651 |
2708 |
10945.00 |
1129.51 |
8.01 |
0.98 |
|
4 |
3816 |
945 |
3654.67 |
669.07 |
6.35 |
0.95 |
|
5 |
16901 |
4799 |
6535.14 |
927.27 |
6.61 |
0.88 |
|
6 |
12507 |
2119 |
2838.34 |
603.14 |
5.81 |
0.90 |
|
7 |
12278 |
2436 |
3009.30 |
866.80 |
7.35 |
0.96 |
|
8 |
8522 |
555 |
757.34 |
245.60 |
4.36 |
0.80 |
|
9 |
26359 |
7354 |
6897.23 |
1011.90 |
7.32 |
0.93 |
|
10 |
3008 |
1514 |
10752.30 |
1290.16 |
8.47 |
0.98 |
|
11 |
12453 |
2035 |
2317.54 |
747.07 |
7.41 |
0.98 |
|
12 |
10182 |
3702 |
7345.52 |
1131.57 |
8.25 |
0.98 |
|
13 |
3061 |
1616 |
10839.00 |
1356.90 |
9.03 |
0.99 |
|
14 |
2557 |
393 |
1515.11 |
387.21 |
4.73 |
0.87 |
|
均值 |
9604 |
2438 |
5354.58 |
846.03 |
6.89 |
0.93 |
|
方差 |
6521 |
1861 |
3547.09 |
329.16 |
1.44 |
0.06 |
2.2样品OTU水平分析
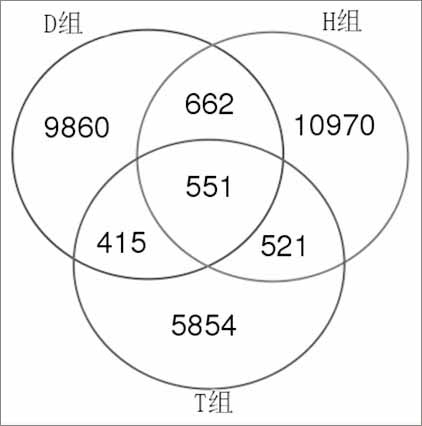
由图2可知,H组共有12704个OTUs,D组共划分了11488个OTUs,T组包含有7341个OTUs。3组样品共有的OTU数目虽仅有551个,但这551个OTU包含了75152条序列,占序列总数的55.90%。共有的OTU数目虽少,但所代表的序列数却超过一半。这部分OTU应该是肥胖人群的“核心OTU”,所代表的微生物类群可能是14个肥胖个体的“核心菌群”,这些细菌在肥胖者肠道中含量较高。将551个共有OTUs通过RDP和Greengene数据库比对发现,其中355个OTU被鉴定为拟杆菌属(Bacteroides),占共有OTU数目的64.43%。包含核心OTUs数目较多的菌属依次是梭菌属(Clostridium,28个OTUs)、栖粪杆菌属(Faecalibacterium,27个OTUs)、考拉杆菌属(Phascolarctobacterium,19个OTUs)和真杆菌属(Eubacterium,17个OTUs)等。说明不同健康状态肥胖人群肠道内的“核心菌属”含量最多的是拟杆菌属。此外,3组样品也存在大量的特有OTU,这部分OTU代表的序列数相对较少,表明不同健康状态肥胖个体的肠道菌群既存在共性也具有特异性。不同人群肠道内是存在共有菌属的,而这些菌属大部分是和机体免疫、代谢、消化等密切相关,在机体肠道内发挥着不可替代的作用。Huse[15]的研究团队对200名人类元基因组计划的志愿者肠道菌群测序数据进行深入挖掘,结果发现200名志愿者有7个共有OTU,被鉴定为栖粪杆菌属、颤杆菌克菌属和拟杆菌属3个菌属,几乎出现在每一个志愿者的肠道中,是200名志愿者的“核心菌群”[15]。
图2 基于OTU水平的Venn图
Fig.2 Venn diagram based on OTU level table
2.3肥胖人群肠道细菌组成
所有样品测序获得的高质量序列共鉴定为9个门类,包括拟杆菌门(Bacteroidetes,54.96%)、厚壁菌门(Firmicutes,38.37%)、变形菌门(Proteobacteria,4.78%)、放线菌门(Actinobacteria,0.037%)、蓝藻菌门(Cyanobacteria,0.078%)、梭杆菌门(Fusobacteria,0.91%)、黏胶球形菌门(Lentisphaerae,0.011%)、无壁菌门(Tenericutes,0.0049%)和疣微菌门(Verrucomicrobia,0.22%)。其中,拟杆菌门和厚壁菌门是绝对的优势菌门,相对含量加和在81.73%~99.09%之间,可见样品中这两大类菌群在人体肠道中的含量丰富。变形菌门的含量次之,平均相对含量为4.78%,其它菌门平均相对含量小于1%,特别是放线菌门的含量小于0.1%。有研究表明健康人体肠道内变形菌门相对含量平均在2.8%左右[16]。本试验调研的肥胖者肠道中变形菌门相对含量较高,这可能是由于受试者中部分人有胃肠道疾病的缘故,已经证实变形菌门的增加和腹泻等肠道炎症性疾病的发生相关[17]。虽然高脂饮食可迅速改变肠道菌群的结构,但对不同个体而言,改变的方向并不相同,共同的结果却都是使得肥胖个体的拟杆菌等高丰度细菌相对含量增加,放线菌门等低丰度细菌的含量减少。
肥胖菌群的相关研究经常用拟杆菌门和厚壁菌门的比值(B/F)作为肥胖菌群的标签,特别是动物模型中肥胖者的肠道菌群B/F值较正常体重低,而这一结论在人体研究中并不一致[18-19]。本研究中,H组的B/F值为1.35,而T组除了14号样品外其它样品厚壁菌门含量均大于拟杆菌门的含量,B/F值最低为0.93(β多样性研究显示14号样品属于离群样品,由此未计算在内)(图4)。本研究发现不是所有肥胖个体的B/F值都是显著降低的,通常在肥胖个体患慢性病时B/F值较单纯肥胖人群低。
属水平,共识别86个菌属,其中拟杆菌属(Bacteroides)占比最高,平均达到44.74%,普氏菌属(Prevotella)相对含量为7.77%,考拉杆菌属(Phascolarctobacterium)相对含量为6.96%,栖粪杆菌属(Faecalibacterium,5.38%)、真杆菌属(Eubacterium,4.26%)、Parasutterella(3.35%)、乳杆菌属(Lactobacillus,3.12%)、瘤胃球菌属(Ruminococcus,2.99%)、梭菌属(Clostridium,2.92%)、小类杆菌属(Dialister,1.73%)、Parabacteroides(1.47%)、巨单胞菌属(Megamonas,1.39%)和罗斯氏菌属(Roseburia,1.35%)的平均相对含量大于1% (图2b)。有研究揭示,肥胖者的肠道中拟杆菌属相对含量高于正常个体[20-23]。同时,也有研究证实,细菌多样性降低往往表现为拟杆菌属的增加,而细菌多样性降低容易引起肥胖,肥胖又是一种潜在的促炎因子,会伴随糖尿病等一些慢性病发生[24]。高脂饮食是很多疾病发生的一个诱因,其主要原因是改变了肠道菌群的结构。
基于16S rRNA基因全长序列,在14个样品中共鉴定到194个细菌种。其中相对含量大于1%的有16个种,大于0.1%的有57个种(图3c)。普通拟杆菌(Bacteroides vulgatus)含量最高为19.32%,人体普氏菌(Prevotella copri)、粪考拉杆菌 (Phascolarctobacterium faecium)、柔嫩梭菌(Faecalibacterium prausnitzii)、直肠真杆菌(Eubacterium rectale)、Parasutterella excrementihominis、单型拟杆菌(Bacteroides uniformis)、Lactobacillus rogosae、四棘蝴蝶鱼拟杆菌(Bacteroides plebeius)、多边型拟杆菌(Bacteroides thetaiotaomicron)等拟杆菌属的一些种和白色瘤胃球菌(Ruminococcus albus)的相对含量大于1%。综上可知,全部样品拟杆菌属的种类最为丰富,由于人体肠道菌群个体差异较大,因此每个样品的优势菌种不同。
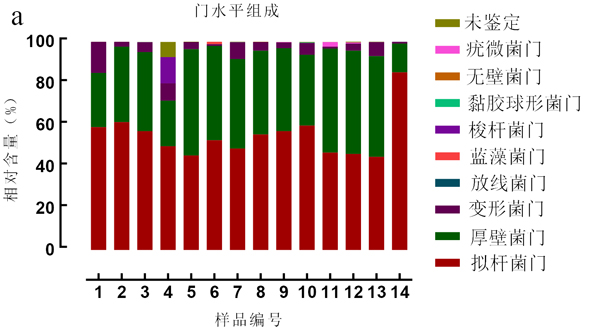
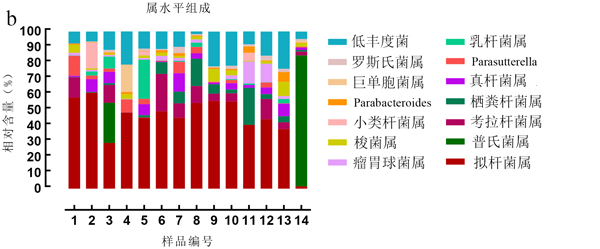
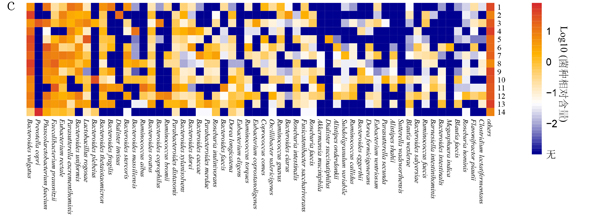
注:a. 门水平组成图;b. 属水平组成图;c. 种水平组成热图(大于0.1%的菌种的相对含量取10的对数值)。
图3肥胖人群肠道细菌组成的相对丰度
Fig.3 Relative abundance of bacterial community of gut bacteria in obese individuals
2.4不同健康状态的肥胖个体肠道细菌比较分析
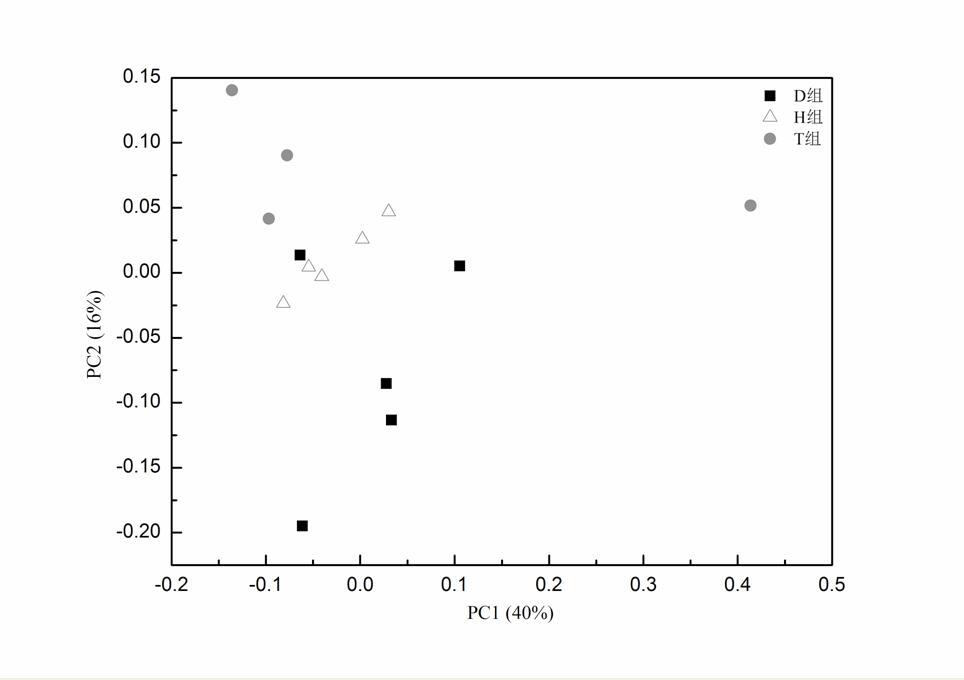
利用基于加权和非加权(weighted和unweighted)的UniFrac距离的主坐标分析(PCoA),分析14个样品细菌结构。基于加权的主坐标分析,其第一主成分和第二主成分的贡献率分别为40% 和16%,虽然有个别交叠现象,但是可以将不同组别区分开。PCoA是研究数据相似性和差异性的方法。本研究中,由图4可知,H组样品除了一个样本,其它和D组、T组距离较远,而T组除了一个离群样本外,其它样本和D组也有分别聚类的趋势。同时,T组中的离群样品是14号样品,14号样品的志愿者同时患有高血压和脂肪肝,可能由于机体患病情况复杂,因此菌群结构和其它样品差异较大。由以上推测,肥胖个体肠道菌群随着健康状态的不同,结构发生了显著改变,而这些改变和患病情况有关。
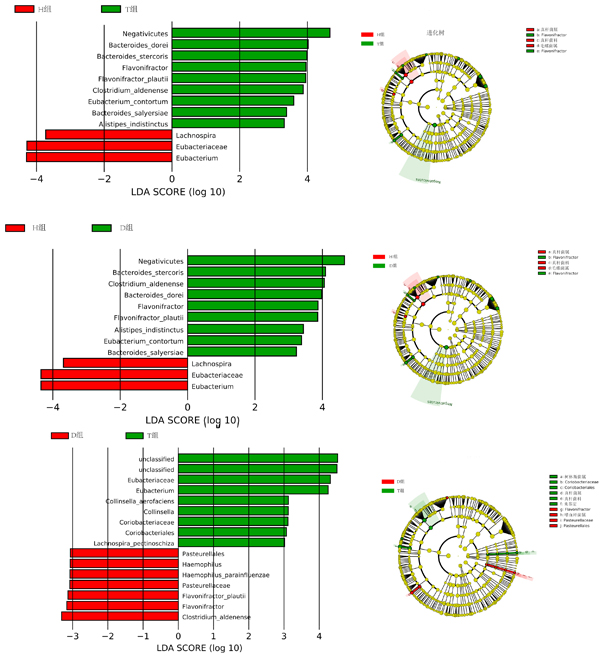
为了进一步研究不同健康状态的肥胖个体肠道菌群的差异,进行基于OUT水平的LEfSe分析(图5)。LEfSe是一种基于线性判别分析(Linear discriminant analysis,LDA)效应量(Effect size)的分析方法,其本质是将非参数统计检验(Kruskal-Wallis 多组检验与 Wilcoxon 秩和检验)与线性判别分析相结合,从而筛选关键的生物标记物种[25]。在种的水平上,取每两组肠道菌群的LDA 差异分析对数得分值大于2的菌种进行比较,结果显示,T组和D组较H组差异菌种均包含7个种,粪便拟杆菌(Bacteroides stercoris)、Clostridium aldenense、Bacteroides dorei、Flavonifractor plautii、Alistipes indistinctus、Bacteroides salyersiae和扭曲真杆菌(Eubacterium contortum)。其中耐胆盐的Alistipes和拟
图4基于加权的UniFrac距离的细菌主坐标分析图
Fig.4 Principal coordinate analysis score plots of bacteria based on the weighted UniFrac distances.
杆菌属的一些菌种显著高于H组。David的研究结果也表明高脂饮食引起这两个菌属的增加[8],然而,进一步发现,当高脂饮食的肥胖人群发生疾病时,这两个菌属显著高于没有疾病表现的单纯肥胖组。此外,拟杆菌属的这3个菌种都是较新归类命名的肠道菌种,如Bacteroides dorei是2006年描述的一个新的种,在临床上的研究较少,2014年西班牙的一个研究团队报道了该菌在乳糜腹泻(慢
注:从上到下3幅图表示每两组的差异物种比较。每个图中,左边图:不同组间LDA评分的分布,X轴表示LDA SCROE(lg),Y轴表示显著不同的粪便细菌种类(LDA SCORE>2);右边图:从内圈到外圈的分类树,其依次显示物种的从属关系,节点大小对应于物种的平均相对丰度,黄色节点表示功能组之间的非显著性差异;红色或绿色区域表明各自代表的分组的差异物种丰度较高。
图5 不同健康状态的肥胖人群肠道细菌LEfSe分析
Fig.5 Analysis of intestinal bacteria in obese individual of different health condition by linear discriminant analysis coupled with effect size (LEfSe)
性肠炎)患者肠道中的相对含量显著增加[26]。T组和D组比较结果显示,T组较D组相对含量显著高的细菌包括真杆菌属、柯林斯菌属和Coribacteriale属下面的菌种Lachnospira peotinoschiza和产气柯林斯菌(Collinsella aerofaciens),其中产气柯林斯菌已被证实和宿主的脂代谢有关,在动脉粥样硬化患者的肠道中被发现富集[27]。这也佐证了本试验的结果:T组是患有高血脂等慢性病的肥胖个体,他们肠道内的产气柯林斯菌属显著高于其它分组,由此解释了高脂饮食导致的肥胖在引发高血压等疾病的同时改变了肠道菌群结构。此外,也有研究表明毛螺菌属(Lachnospira)的菌种可能和II型糖尿病有相关性[28]。D组较T组相对含量高的菌种包括副流感嗜血杆菌(Haemophilus parainfluenzae)、Clostridium aldenen和Flavonifractor plautii。其中副流感嗜血杆菌是致病菌,该菌可引起肾炎、腹膜炎等多种炎症性疾病[29]。而Flavonifractor plautii在一项关于日本肥胖人群肠道菌群的研究中发现,该菌在肥胖者肠道内的相对含量低于正常体重的人群[30]。本试验进一步研究发现该菌在患不同疾病的肥胖人群间的含量也存在显著差异,慢性病组(T组)低于胃肠道疾病组(D组)。总之,疾病影响着肥胖人群肠道细菌的结构。
3.结论
在过去的几年里,全球肥胖发病率至少上升了7倍,肥胖已经成为世界性问题。研究已经表明,高脂饮食导致肥胖的同时改变了肠道菌群的结构,而肥胖患者存在很多健康隐患,例如肥胖会导致的胰岛素抗性是II型糖尿病患者增加的驱动力,全球超过80%的糖尿病患者超重。本试验研究高脂饮食导致的肥胖个体肠道菌群结构的同时,比较了不同健康状态的肥胖患者肠道菌群的特点。研究发现,高脂饮食的肥胖个体的肠道内,拟杆菌属在其核心菌属中占比最高,同时肥胖导致菌群多样性降低,优势菌占比更高,低丰度细菌的占比更低。此外,肥胖者的肠道菌群结构也存在个体差异,而这些差异很可能和肥胖者的不同并发症有关。研究发现患有慢性病的肥胖者,其肠道菌群的B/F值显著低于未患病的肥胖个体,同时柯林斯菌和毛螺菌科的菌种显著高于未患慢性病的肥胖者。此外,患有腹泻等胃肠道疾病的肥胖个体,同时表现出变形菌门下的一些条件致病菌的增加。总之,肥胖个体的肠道菌群的多样性受机体健康状态的影响,肥胖引起的并发症可能和肠道菌群的改变相关。
参考文献
[1] TREMAROLI V,BACKHED F. Functional interactions between the gut microbiota and host metabolism[J]. Nature,2012,489(7415):242-249.
[2] TSAI F,COYLE W J. The microbiome and obesity: is obesity linked to our gut flora?[J]. Current Gastroenterology Reports,2009,11(4):307-313.
[3] NICHOLSON J K,HOLMES E,KINROSS J,et al. Host-gut microbiota metabolic interactions [J]. Science,2012,336(6086):1262-1267.
[4] QIN J,LI Y,CAI Z,et al. A metagenome-wide association study of gut microbiota in type 2 diabetes[J]. Nature,2012,490(7418):55–60.
[5] TURNBAUGH P J,HAMADY M,YATSUNENKO T,et al. A core gut microbiome in obese and lean twins[J]. Nature,2009,457(7228):480–484.
[6] RIDAURA V K,FAITH J J,REY F E. Gut microbiota from twins discordant for obesity modulate metabolism in mice[J]. Science,2013,341(6150):1079-1089.
[7] GILL S R, POP M,DEBOYRT,et al. Metagenomic analysis of the human distal gut microbiome[J]. Science,2006,312(5778):1355-1359.
[8] DAVID L A, MAURICE C F,CARMODY R N,et al. Diet rapidly and reproducibly alters the human gut microbiome[J]. Nature,2014,585(505):559-563.
[9] LEY R E,TURNBAUGH P J,KLEIN S,et al. Microbial ecology: human gut microbes associated with obesity[J]. Nature,2006,444(7122):1022-1023.
[10]DUNCAN S H,LOBLEY G E,HOLTROP G,et al. Human colonic microbiota associated with diet, obesity and weight loss[J]. International Journal of Obesity,2008,32(11):1720-1724.
[11]THAISS C A,ZMORA N,LEVY M,et al. The microbiome and innate immunity[J]. Nature, 2016,535(7610):65–74.
[12]MOSHER J J,BERNBERG E L,SHEVCHENKO O,et al. Efficacy of a 3rd generation high-throughput sequencing platform for analyses of 16s rRNA genes from environmental samples[J]. Journal of Microbiological Methods,2013,95(2):175–181.
[13]KIM M,OH H S,PARK S C,et al. Towards a taxonomic coherence between average nucleotide identity and 16s rRNA sequence similarity for species demarcation of prokaryotes[J]. International Journal of Systematic and Evolutionary Microbiology,2014,64(Pt2):346–351.
[14]LI J,XU H Y,SUN Z H,et al. Effect of dietary interventions on the intestinal microbiota of Mongolian hosts[J]. Science Bulletin,2016,20(61):1605-1614.
[15]HUSE S M,YE Y,ZHOU Y,et al. A core human microbiome as viewed through 16S rRNA sequence clusters[J]. PLoS One,2012,7(6): e34242.
[16]PARK S H,KIM K A,AHN Y E,et al. Comparative analysis of gut microbiota in elderly people of urbanized towns and longevity villages[J]. BMC Microbiology 2015,15(49):1-9.
[17]WONG J M,SOUZA W R, DE KENDALL C W C,et al. Colonic health: fermentation and short chain fatty acids[J]. J Clin Gastroenterol,2006,40(3):235–243.
[18]LEY R E,BÄCKHED F,TURNBAUGH P,et al. Obesity alters gut microbial ecology[J]. PNAS,31(102):11070–11075.
[19]FINUCANE M M, SHARPTON T J, LAURENT T J, et al. A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter[J]. PLoS One,2014,9(1): e84689.
[20]DUNCAN S H,LOBLEY G E,HOLTROP G,et al. Human colonic microbiota associated with diet, obesity and weight loss[J]. International Journal of Obesity,2008,32(11):1720–1724.
[21]SCHWIERTZ A,TARAS D,SCHAFER K,et al. Microbiota and SCFA in lean and overweight healthy subjects[J]. Obesity,2010,18(1):190–195.
[22]WU G D, CHEN J, HOFFMANN C, et al. Linking long-term dietary patterns with gut microbial enterotypes[J]. Science,2011,334(6052):105–108.
[23]DE FILIPPO C, CAVALIERI D, DI PAOLA M, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa[J]. Proceedings of the National Academy of Sciences of the United States of America,2010,107(33):14691–14696.
[24]TILG H,MOSCHEN A R. Microbiota and diabetes: an evolving relationship[J]. Gut,2014,63(9):1–9.
[25]WU X Y,ZHANG H X,ZHANG H H,et al. Analysis and comparison of the wolf microbiome under different environmental factors using three different data of Next Generation Sequencing[J]. Scientific Reports,2017,7(1):11332.
[26]NCHEZ E S,DONAT E,RIBES-KONINCKX C,et al. Intestinal Bacteroides species associated with coeliac disease[J]. J Clin Pathol,2010,63(63):1105-1111.
[27]XU J,XU T,BU X Q,et al. The predictive value of waist-to-height ratio for ischemic stroke in a population-based prospective cohort study among Mongolian men in China[J]. PloS One,9(10):e110245.
[28]KAMEYAMA K,ITOH K. Intestinal colonization by a Lachnospiraceae bacterium contributes to the development of diabetes in obese mice[J]. Microbes Environ,2014,29 (4):427-430.
[29]官振标. 慢性阻塞性肺疾病中副流感嗜血杆菌感染的临床和致病性研究[D]. 上海:第二军医大学,2010.
[30]KASAI C,SUGIMOT K. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing[J]. BMC Gastroenterology,2015,15(1): 100.
 手机浏览
手机浏览